
05240 Eigenherstellung von Medizinprodukten und In-vitro Diagnostika im Krankenhaus und die daraus folgenden (Dokumentations-)Pflichten
|
Das Medizinprodukterecht-Durchführungsgesetz – MPDG von 2020 setzt der Europäischen Rechtsgrundlage definiert in der MDR bzw. IvDR keine weiteren Forderungen für die so genannte Eigenherstellung zur Seite. Daher können wir uns im Folgenden auf die Forderungen der Europäischen Rechtsdokumente beschränken und betrachten, welche Konsequenzen die Eigenherstellung auf das QM-System eines Krankenhauses, einer Pflegeeinrichtung oder auf eine Arztpraxis hat. Diese Konsequenzen für die Leistungserbringer und die mögliche Realisation, sollen hier kurz beleuchtet werden. Der Praxisbezug steht dabei immer klar im Vordergrund. Es gilt zu beachten:
Arbeitshilfen: von: |
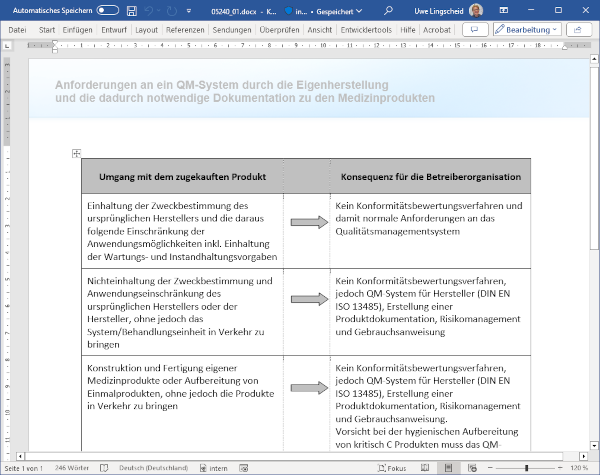
1 Wenn das Krankenhaus zum Hersteller wird
CE-Kennzeichnung
Längst ist bekannt, dass Herstellerunternehmen von Medizinprodukten, einschließlich In-vitro-Diagnostika, entsprechend dem deutschen Medizinprodukterecht-Durchführungsgesetz bzw. den Europäischen Verordnungen vor dem erstmaligen Inverkehrbringen bzw. Inbetriebnehmen eine CE-Kennzeichnung auf dem Produkt anbringen müssen. Diese Kennzeichnung steht für die Übereinstimmung des jeweiligen Produkts mit den grundlegenden Sicherheits- und Leistungsanforderungen der geltenden Europäischen Verordnung, d. h., der Hersteller bestätigt hierdurch die Einhaltung der Produktanforderungen der Rechtslage bezüglich Produktsicherheit und auch Leistung zum Zeitpunkt der Auslieferung bzw. vor dem ersten Einsatz durch den Anwender und über die gesamte Produktlebenszeit.
Längst ist bekannt, dass Herstellerunternehmen von Medizinprodukten, einschließlich In-vitro-Diagnostika, entsprechend dem deutschen Medizinprodukterecht-Durchführungsgesetz bzw. den Europäischen Verordnungen vor dem erstmaligen Inverkehrbringen bzw. Inbetriebnehmen eine CE-Kennzeichnung auf dem Produkt anbringen müssen. Diese Kennzeichnung steht für die Übereinstimmung des jeweiligen Produkts mit den grundlegenden Sicherheits- und Leistungsanforderungen der geltenden Europäischen Verordnung, d. h., der Hersteller bestätigt hierdurch die Einhaltung der Produktanforderungen der Rechtslage bezüglich Produktsicherheit und auch Leistung zum Zeitpunkt der Auslieferung bzw. vor dem ersten Einsatz durch den Anwender und über die gesamte Produktlebenszeit.
Mit der Forderung zur CE-Kennzeichnung ergeht zeitgleich die Notwendigkeit der Errichtung und Aufrechterhaltung eines QM-Systems, das je nach Wahl des möglichen Konformitätsbewertungsverfahrens extern gegenüber einer Benannten Stelle nachweispflichtig ist. Jedoch auch die Hersteller von Produkten mit geringem Risiko (Medizinprodukte der Klasse 1 oder IvD der Klasse A) sind in der Pflicht der Einführung und Erhalt eines dokumentierten QM-Systems, das statistisch von den Behörden kontrolliert wird. Daneben gibt es selbstverständlich noch die Forderung der Haltung einer Produktdokumentation.
